
Differential Expression Analysis with RNA-Seq:
A Step-By-Step Guide
In this step-by-step guide, you will perform an RNA-Seq differential expression analysis from start (raw FASTQ files) to finish (figures summarizing differential expression and list of differentially expressed genes). The files, apps, and workflow are pre-loaded as part of a public project hosted on the CGC titled “Bulk RNA-Seq Transcription Profiling of HSV-1 Infected Hepatocellular Carcinoma Cells” (login required). The data were originally published in Mani SKK, et al., (2020).
This tutorial is intended for a user with little to no experience with the Cancer Genomics Cloud or cloud-based computing, and who may or may not have experience with performing RNA-Seq analysis. If you already feel comfortable with performing analyses on the CGC, a succinct overview of the below procedure is available on the CGC Knowledge Center.
You must have an account on the CGC and compute credits attached to that account. Pilots funds are available through the NCI; contact Support to request them. Performing the below analysis will take approximately 1.5 hours and will cost between $0.50 - $1.50.
Table of Contents
Getting Started With the Public Project
On the CGC and other Seven Bridges platforms, work is organized as projects, which contain files, apps, and tasks. For an in-depth discussion of these terms, see “Helpful terms to know” in the comprehensive user guide.
Here, we will start with a fully formed project and walk through each of the parts. You will learn how to copy a public project, explore the files and apps available within it, run the associated analysis, and see the results.
Find and copy the public project
From your user dashboard, click on the Public Projects dropdown menu and find the public project titled “Bulk RNA-Seq Transcription Profiling of HSV-1 Infected Hepatocellular Carcinoma Cells”. Click the project name.
Find the information tooltip next to the project title at the top of the page. Click the tooltip, and find the green Copy Project button.
Copy the project to your workspace.
Here’s what performing these steps looks like:

Animation showing the steps needed to find and copy the public project to your own workspace.
Explore the Preloaded Files
From the project dashboard, click the Files tab at the top of the page to go to the Files view.
There are three folders in the file browser: Input, Output, and Reference.
Click on the Input folder to see the raw data files used in this project.

The files view offers a search function, in addition to a number of ways to filter the files that you see in the listing. The default dropdowns in this view are Extension, Sample ID, Task ID, and Tag. However, you can also add a filter for any metadata that is associated with any of the files.
Filtering files

Click the gray plus button to open a dropdown of all available filters for these files.
Scroll down to near the bottom, and add Genotype as a filter.
The new Genotype filter will display its available options.
After adding the new filter, we can see that there are twelve WT files and eighteen KD files in the set. These correspond to six WT samples and nine KD samples, because there are two paired FASTQ read files for each sample; the study used paired-end sequencing data.
This metadata is unique to this particular data set, and will be used in the differential expression workflow.
Navigating files
There are a number of ways to navigate through the files associated with a project, but when files are organized into folders as they are in this project, one convenient way is to enable the sidebar:
Click the sidebar icon next to the word “Files” right above the search bar.
“Output” has a preview of the output files that will be produced by the workflow.
“Reference” holds the genome reference file and gene annotation file.

Metadata
Metadata is an important aspect of files on the Cancer Genomics Cloud. These steps aren’t shown because they’re not required for the analysis, but we encourage you to explore the file metadata a little:
Try looking at the file GRCh38ERCC.ensembl95.gtf in the Reference folder by clicking its name.
Scroll all the way down to the bottom of the metadata page to view information about how this reference file was created.
Next, we’ll look at the workflow that you will use with these files.
Explore the Differential Expression Workflow
Click on the word Apps in the project header to view the software tools associated with the project. For this example, there is only one listed, and it is a workflow - a chain of software connected with inputs and outputs.

The Apps view in the Bulk RNA-Seq project
Click on the name of the workflow Differential Expression - Salmon + DESeq2 and view its diagram and description.

Workflow diagram
The diagram of the workflow provides an overview of everything that is going to happen once we run this workflow in a task. Apps (software) are shown in teal circles, and inputs and (files) are shown in gray circles.
This diagram has been rearranged to make certain aspects clearer, but the connections and apps represented are identical to the one you see at the top of the workflow page.
Notice that the software FastQC will be run on each of the FASTQ read files. A more typical RNA-Seq workflow would likely involve running FastQC by itself to check the quality of the input FASTQ read files. Here, the FASTQ read files are known quantities, but we’re running FastQC anyway to be able to demonstrate what its output looks like.
The FASTQ read files are being piped directly into the Salmon workflow. Because FastQC and Salmon are independent from each other, those two apps will run roughly simultaneously. The app DESeq2, however, relies on an input from Salmon—the transcript-level quantification—so DESeq2 will not run until after Salmon has completed for each sample.
-
This workflow uses quasi-mapping (Salmon) rather than sequence alignment to count transcripts. In alignment-based methods (e.g., Bowtie and BWA), trimming reads helps you keep good reads that would have trouble aligning because of low quality or adapter sequence. In quasi-mapping methods, bits of sequence that aren't found in the reference are simply not counted, so trimming is typically not necessary.
Workflow description
The Description of the workflow provides additional information about what the workflow does and how to use it. On the right hand side, under Basic Information, you will see information about the workflow itself; you should see that your username appears next to Contributors, because this workflow is a copy that you have imported into your workspace. Workflow descriptions usually include budgeting and benchmarking information that estimates how long a workflow will take to run and how much it might cost, depending on the size and number of input files.
Ports: defining the workflow requirements
Scroll down to the Ports section of the page. This shows what kinds of files are used as inputs, any special app settings that should be used, and what the output files will be.
Next to each row under ports is a tooltip icon that looks like a question mark in a black circle.
Click on App Settings, and then move your mouse cursor over the tooltip next to factor (“Covariate of interest”).

The tooltip’s text for factor says: “The samples will be grouped according to the chosen variable of interest. This needs to match either a column name in the provided phenotype data CSV file or a metadata key. If the latter is true, then all the input files need to have this metadata field populated.”
What does this mean?
This means that in order to use DESeq2 for differential expression analysis a variable has to be defined to tell the software how the samples are different from each other (i.e., phenotypes or clinical traits). We can provide the DESeq2 app with a text document that defines phenotype data, but the tooltip text is telling us that DESeq2 will be able to get that information from the metadata attached to each input file. We’ll explain this in greater detail below, when you set the workflow up to run.
Run the Workflow Yourself
So that you can see how all of these pieces (files, apps, and tasks) are put together, we’re going to run the workflow from start to finish.
Specify input files
Go to the Apps tab and click the green Run button. This creates a new “draft” task.
Click Select Files and you will be taken to a Files view.
Notice that the Extension filter drop down is bolded, and says that there are 4 extensions selected. This is because the workflow is informed about the kind of input files it requires (through ports), and so the files view pre-filters based on file extension.

The input files are automatically filtered to FASTQ files based on the workflow’s settings.
At the top left of the file listing pane, find the checkbox with a drop down arrow.
Click the arrow, and click “Select All.”
Click the blue Save selection button at the top right of the page.
You will be returned to the draft task view with the FASTQ read files added.

Selecting the FASTQ files and clicking “Save Selection” returns you to the task with input files completed.
Scroll down in the draft Task view and perform the same steps for the input ports GTF annotation files and Transcript FASTA or Salmon Index files—the other two inputs that have red text telling us that they are required. For the GTF annotation file, there will only be one file available to select.
The Salmon Index, on the other hand, requires a TAR file extension, and there are several other TAR files in this project that are not actually index files. In this project, we have used a tag to identify which TAR file is actually an index file.
Click the Tags filter dropdown, and select the tag index.
Now only one file remains, so select it and add it to the draft task as before.

Both the index file and some of our outputs have TAR extensions, so we used tags to uniquely identify the index file.
DESeq2 App Settings
Now that all the input files are taken care of, you need to add App Settings. The app in this workflow that performs differential expression is an implementation of DESeq2, a powerful package that is part of the R programming language. The Seven Bridges DESeq2 app is set up to make comparisons between samples based on information in the metadata. Specifically, DESeq2 needs to know your covariate of interest (which should be one of the metadata fields for the FASTQ files), and then needs to know what value to use as the reference (e.g., the control), and what value to use for test (e.g., the experimental sample).
Here is where we can put to good use the Genotype metadata information we learned about above in the Explore the Preloaded Files section.
For Covariate of interest, type in the word “Genotype”
For Factor level - reference, type in “WT” (for wild-type)
For Factor level - test, type in “KD”
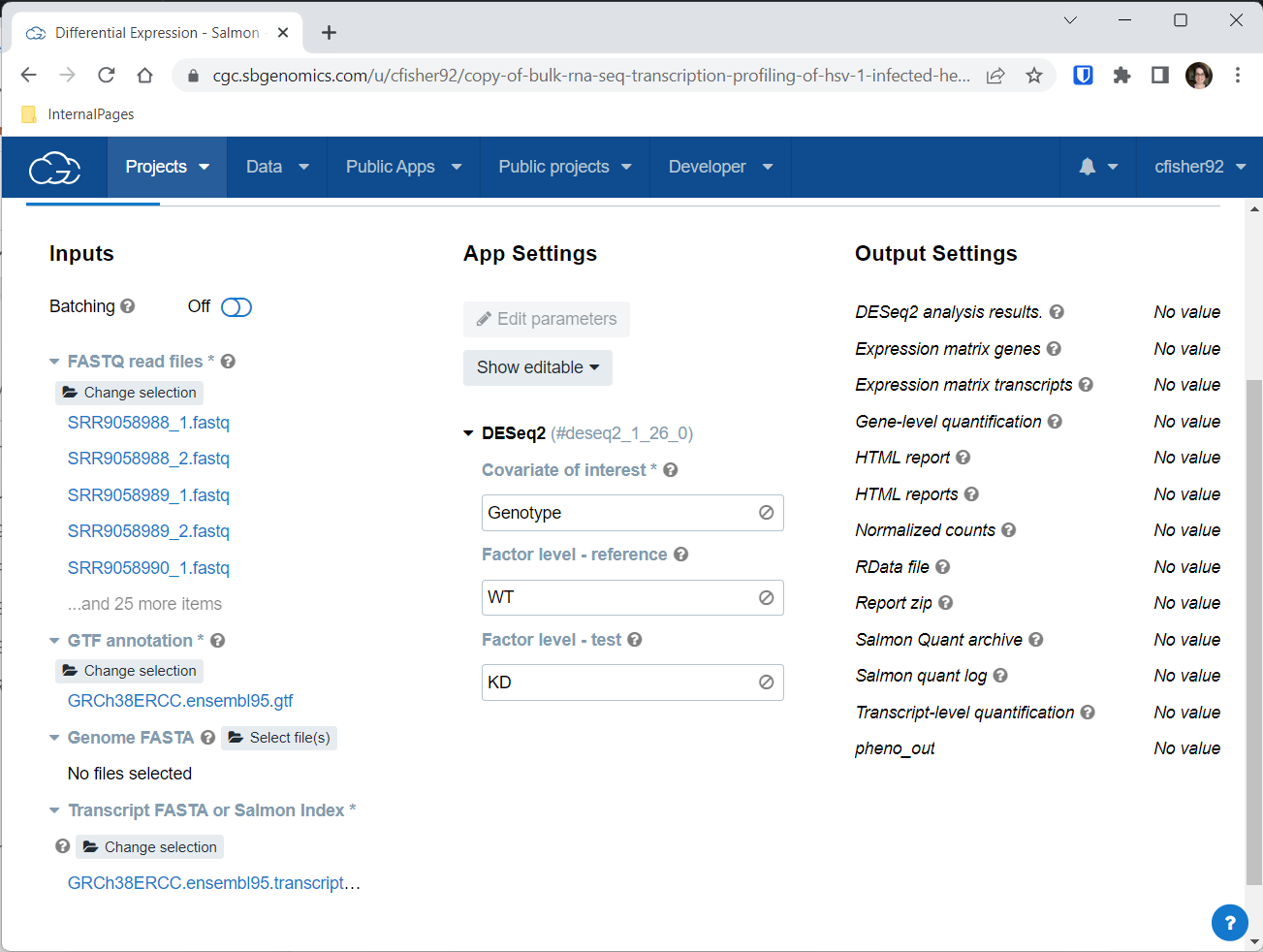
The completed task view, showing the entries under App Settings for covariate of interest, factor level - reference, and factor level - test.
Output Settings
Under the header Output Settings, there is a list describing all of the files that will be created as a result of running this task. Use the tooltip icons to learn more about each of these output files.
Nota bene: FastQC will produce HTML reports for each input FASTQ read file, and these are called “HTML reports” by the workflow. The Seven Bridges DESeq2 App also produces an HTML report, and this is called “HTML report.” Prior to running the task, these seem redundant, but after the task has completed, it is much clearer which is which!
Run the Task
So far, nothing we have done, including copying files over to your workspace, has incurred any compute time costs. But now, we’re ready to hit the Run button and actually use some cloud computing resources. You should have pilot funds available for this part of the tutorial; if you do not, please contact our Support Team (support@sbgenomics.com) and request pilot funds.
When you click the blue Run button, the task will be queued for a few moments, and then you will see that it has started running after the cloud platform finds available resources.

This is how the task view will change as the workflow is set to run (1), gets queued (2), and finally is running (3).
The task will normally take between 45 minutes to 2 hours and 15 minutes to complete, as we have set it up here. When I ran the task for this tutorial, it took 1:56 hours, for a total compute cost of $1.18.
-
You’ll receive an email to the address you registered with, informing you that the task has completed. (If anything went wrong, you’ll be alerted to that by email, too!)
View the Results
Once the task is completed, the Task tag will change to green Completed status, and the output settings will be populated with links to each of the output files. Return to the task view (either by following the link from your email, or by navigating to Tasks from your project dashboard) and you will see something like the below:

There’s a lot to unpack here! The workflow has saved a comma-separated value file of DESeq2 analysis results, two expression matrices (gene level and transcript level), the Salmon output for each of the 30 samples, and many more files besides. Let’s zero in on the main summary produced by this workflow under HTML report.
HTML Report
Scroll down in the completed task view to find the HTML report entry.
Click the filename that appears underneath it, which should start with “SRR905.DEAnalysis.deseq2…”
That link leads to the file page for the report, but opens onto a Preview pane that displays the HTML rendered output.
Click Expand Report to open the HTML report in a new tab.
From here, you can scroll through some summary figures and text about the differential expression results. Of particular interest may be the PCA plot, showing how different samples cluster together based on differential expression; the cluster dendrogram, which shows in a hierarchical fashion which samples’ gene expression profiles are most similar to each other; and the text Analysis Summary, which details how many genes have higher or lower expression in genotype KD relative to genotype WT. While none of these would be considered final results, these figures present an exploratory analysis that would form the basis for further analysis of the differentially expressed genes.
Tabular Results by Gene
Return to the completed task view, and scroll down to find the output under the heading DESeq analysis results.
Click on the filename to open the file preview.
The default sorting is alphabetical by gene name. The first several genes had counts of 0, so the first results you see are NAs!
Click on the sorting arrows to sort the preview by different fields. Try sorting the results by padj (the false discovery rate) ascending, to put the smallest adjusted p-values at the top.
This file, the DEAnalysis.out.csv file, is the raw results of the differential expression analysis. It can be downloaded directly to your computer by clicking the Download button, and, if you wish, could be opened in a spreadsheet program, though this will incur file transfer costs, and is strongly NOT recommended because of spreadsheet software auto-correct features that change some gene names to dates. More commonly, this tabular results file is used as the input for software like R or SAS for creating graphs and analyzing relationships between variables. Even for these purposes, downloading is not necessary, because you can perform your R analysis in Data Studio.
Data studio
Your CGC project has a final tab that we haven’t looked at yet: Data Studio. The Data Studio allows interactive exploration and analysis of your data on the cloud via Python Jupyter Notebooks, R and RStudio, or (coming soon!) SAS Studio. This public project contains a saved session of Data Studio to provide an example of how this works.
Click the Data Studio tab at the top of the page.
This page lists the Data Studio analyses that have been run. Click the name of the saved session, “Copy of DESeq2 Visualization.”
The session overview page shows the files that are part of the Data Studio workspace, including a few .png files of figures created during the session. You can click those to preview them.
Click the blue Start button to open RStudio in a cloud instance. This will incur computational costs, on the order of about $0.34 per hour. Costs can be viewed by click Settings on the session overview page.
Starting up the instance will take 3 to 5 minutes as the resources are secured and software is loaded.
Once the session is ready, an RStudio environment will be loaded onto your screen. If you are familiar with R and RStudio, you can navigate this the way you would on your own computer. Click the File menu and select Open to browse the files attached to the session. The file rna_visualization.R contains a script that explores the DESeq2 results and produces the two figures below.

Figure 1 A volcano plot showing significantly upregulated and significantly downregulated genes from the results of this analysis.

Figure 2 A plot of expression values for nine genes across all samples, comparing samples with the KD genotype to samples with the WT genotype.
-
Check the documentation at https://docs.cancergenomicscloud.org/docs/rstudio-quick-reference for a general guide, and learn more about the Data Studio analysis environment here: https://docs.cancergenomicscloud.org/docs/about-data-cruncher
Extension: Repeat the workflow using different test variables
Differential expression analysis uses comparisons across data to understand changes in gene expression related to clinical traits. In this walkthrough, we used the genotype trait as the covariate of interest, and its two values, WT and KD, as the reference and test conditions. The public project dataset has another trait that can be used in this way—HBV+ or HBV- infection status.
As an extension of this walkthrough, try doing the following:
Examine the metadata of the input files to find HBV status metadata by going to the Files View.
Filter by Extension, selecting FASTQ
Click the plus button to add a new filter, induced_hbv_replication
Note the two values available, HBV_neg and HBV_pos.
Create a new task by going to the completed task and click the blue Edit and rerun button.
Replace the covariate of interest value with “induced_hbv_replication”
Replace the Factor level - reference with “HBV_neg”
Replace the Factor level - test with “HBV_pos”
This will create a new analysis of the same data, but this time, the differential expression comparison will be made across HBV infection status rather than genotype. Run the task with these new settings and compare the resulting HTML Report to the first one.
Analyze your own data using this workflow
To repeat this workflow with your own data, you will need to do the following:
Pull your input files (FASTQ reads) into your project using any of the methods discussed in this tutorial:
Bring Your Data to the CGCAdd metadata to your files that identifies their paired-end status (if applicable) and distinguishes the control samples (i.e., the DESeq2 reference) from the experimental samples (the DESeq2 test). Learn more about how to add metadata to your files here:
Metadata for your private dataSet up your DESeq2 App Settings to use your metadata key as the covariate of interest and the two values you selected for reference and test.